
The Kossiakoff Group’s research interests are to provide a molecular understanding of how molecular recognition governs virtually all aspects of biological function. To study these issues our group employs a combination of X-ray crystallography and cryo-EM, site-directed mutagenesis, phage display and biophysical analysis. The Kossiakoff group has also pioneered a new technology called “chaperone-assisted” crystallography, which has facilitated the structural analyses of protein systems that had been totally recalcitrant to other approaches. The group has also been at the forefront of developing synthetic antibodies. These synthetic antibodies are much more powerful than traditional monoclonal antibodies and have the potential to completely replace them for uses in live cell imaging and proteomics.
Latest Publications

Slezak T; O'Leary K M; Avella T G; Musial N; Li J; Andrzejczak A; Scott E F; Le D A; Kossiakoff A A
Dynamic translocation of Inside-Out proteins to the cell surface underlies cellular adaptation to cancer-induced stress Journal Article
In: Proc Natl Acad Sci U S A, vol. 123, no. 13, pp. e2529493123, 2026, ISSN: 1091-6490.
@article{pmid41875162,
title = {Dynamic translocation of Inside-Out proteins to the cell surface underlies cellular adaptation to cancer-induced stress},
author = {Tomasz Slezak and Kelly M O'Leary and Tanya Guevara Avella and Natalia Musial and Jinyang Li and Anna Andrzejczak and Elizabeth F Scott and Duc Anh Le and Anthony A Kossiakoff},
doi = {10.1073/pnas.2529493123},
issn = {1091-6490},
year = {2026},
date = {2026-03-01},
urldate = {2026-03-01},
journal = {Proc Natl Acad Sci U S A},
volume = {123},
number = {13},
pages = {e2529493123},
abstract = {Inside-Out (I-O) protein display, the noncanonical surface localization of intracellular proteins, represents an underexplored feature of tumor cell biology. Here, we map the molecular landscape and trafficking mechanisms that control the presentation of I-O proteins on cancer cell membranes. Employing APEX2-mediated proximity biotinylation and a custom antibody generation and validation platform, we identified approximately 140 high-confidence I-O proteins, primarily ribosomal, proteasomal, chaperone, and translation factors, notably enriched in protein families associated with stress-response pathways. Validation of 500 antibodies encompassing 40 I-O targets across seven tumor cell lines confirmed selective and robust surface localization, while in vivo imaging in mouse xenografts demonstrated pronounced and tumor-specific antibody accumulation. I-O proteins were absent on peripheral blood mononuclear cells (PBMCs) and in normal tissues, indicating cancer cell selectivity. Functional analyses revealed that I-O protein tethering to the membrane is dependent on heparan sulfate interactions; enzymatic removal of these glycans led to the clearance of I-O proteins from the cell surface. Notably, the removed proteins returned to baseline levels within 6 h, indicating a dynamic balance related to Endoplasmic Reticulum (ER)-Golgi trafficking and cellular stress. Nearly half of these I-O proteins overlapped with known stress granule (SG) components; however, stress elements that promote SG formation do not similarly affect surface display of I-O proteins. Furthermore, I-O proteins are present on standard cancer cell lines under lower stress levels needed to induce SG formation, suggesting parallel yet mechanistically distinct aspects of the stress response. These findings position I-O display as a paradigm in protein trafficking, different from traditional secretion pathways and closely linked to stress response.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Alexander J A N; Chen S; Mukherjee S; de Capitani M; Irobalieva R N; Rossi L; Agrawal P; Kowal J; Meirelles M A; Aebi M; Reymond J; Kossiakoff A A; Riniker S; Locher K P
Structures of ALG3/9/12 reveal the assembly logic of the N-glycan oligomannose core Journal Article
In: Nat Chem Biol, 2026, ISSN: 1552-4469.
@article{pmid41807832,
title = {Structures of ALG3/9/12 reveal the assembly logic of the N-glycan oligomannose core},
author = {J Andrew N Alexander and Shu-Yu Chen and Somnath Mukherjee and Mario de Capitani and Rossitza N Irobalieva and Lorenzo Rossi and Parth Agrawal and Julia Kowal and Matheus A Meirelles and Markus Aebi and Jean-Louis Reymond and Anthony A Kossiakoff and Sereina Riniker and Kaspar P Locher},
doi = {10.1038/s41589-026-02164-7},
issn = {1552-4469},
year = {2026},
date = {2026-03-01},
urldate = {2026-03-01},
journal = {Nat Chem Biol},
abstract = {Asparagine-linked glycans are essential for the maturation and function of most eukaryotic secretory proteins. The biosynthesis and transfer of dolichylpyrophosphate-anchored GlcNAcManGlc glycan is a highly conserved process occurring in the endoplasmic reticulum (ER) membrane and involving over a dozen membrane proteins whose dysfunction is linked to congenital disorders of glycosylation (CDGs). Three membrane-integral mannosyltransferases, ALG3, ALG9 and ALG12, mediate four consecutive mannosylation reactions that convert GlcNAcMan to GlcNAcMan. Here, using chemoenzymatically synthesized lipid-linked glycan donor and acceptor analogs, we recapitulated this biosynthetic pathway in vitro. High-resolution cryo-electron microscopy structures of pseudo-Michaelis complexes of each step revealed how the branched glycan is accurately synthesized and unwanted side products are averted. Molecular dynamics simulations and mutagenesis studies uncovered a subtle but precise mechanism selecting the dolichylphosphomannose donor substrate over dolichylphosphoglucose, which is also present in the ER membrane. Our results also provide mechanistic explanations for enzyme dysfunction in CDGs and offer opportunities for N-glycan engineering.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Erramilli S K; Nosol K; Pietrzak-Lichwa K; Schmandt N; Li T; Tokarz P; Hou J; Zhao M; Perozo E; Kossiakoff A A
Conformational ensembles of the magnesium channel CorA reveal structural basis for channel gating Journal Article
In: Proc Natl Acad Sci U S A, vol. 123, no. 8, pp. e2512532123, 2026, ISSN: 1091-6490.
@article{pmid41701836,
title = {Conformational ensembles of the magnesium channel CorA reveal structural basis for channel gating},
author = {Satchal K Erramilli and Kamil Nosol and Krzysztof Pietrzak-Lichwa and Nicolaus Schmandt and Tian Li and Piotr Tokarz and Jingkai Hou and Minglei Zhao and Eduardo Perozo and Anthony A Kossiakoff},
doi = {10.1073/pnas.2512532123},
issn = {1091-6490},
year = {2026},
date = {2026-02-01},
urldate = {2026-02-01},
journal = {Proc Natl Acad Sci U S A},
volume = {123},
number = {8},
pages = {e2512532123},
abstract = {In prokaryotes, CorA is the primary influx pathway for magnesium, a critical divalent cation in cellular physiology and biochemistry. Mechanistic studies show that homopentameric CorA is regulated through an intracellular [Mg]-dependent negative feedback loop, involving the asymmetric participation of individual subunits. To understand the connection between asymmetry and activation, we used single-particle cryo-EM to solve sixteen structures of nanodisc-reconstituted CorA. We utilized conformation-specific synthetic antibodies to stabilize subtle but significant conformational differences in the cryo-EM structures. Our results demonstrate that CorA exists as a set of conformational ensembles, where population size inversely correlates with intracellular Mg concentration. These ensembles include channels with a variety of pore conformations, both constricted and dilated, suggesting a spectrum of active CorA functional states. The ensembles connect asymmetric structural transitions in the cytoplasmic domain with conformational changes in the permeation pathway via an electrostatic network, ultimately controlling channel-gating events. We believe that these results establish a framework for understanding magnesium homeostasis in prokaryotic systems.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
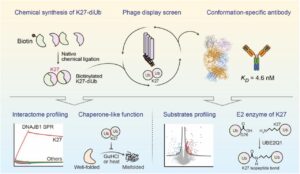
Han C; Weng Y; Zheng Q; Qu Q; Erramilli S K; Su Z; Duan Y; Han Y; Zhai X; Li J; Kossiakoff A A; Pan M; Zhao M; Liu L; Yu Y
Conformation-specific Antibody Deciphers K27-linked Ubiquitination in Chaperone-Mediated Proteostasis Journal Article
In: bioRxiv, 2025, ISSN: 2692-8205.
@article{pmid41446272,
title = {Conformation-specific Antibody Deciphers K27-linked Ubiquitination in Chaperone-Mediated Proteostasis},
author = {Chengxiao Han and Yicheng Weng and Qingyun Zheng and Qian Qu and Satchal K Erramilli and Zhen Su and Yujuan Duan and Yunxi Han and Xiaoguo Zhai and Jingxian Li and Anthony A Kossiakoff and Man Pan and Minglei Zhao and Lei Liu and Yuanyuan Yu},
doi = {10.64898/2025.12.18.695067},
issn = {2692-8205},
year = {2025},
date = {2025-12-01},
urldate = {2025-12-01},
journal = {bioRxiv},
abstract = {Lysine 27 (K27)-linked polyubiquitination plays critical yet incompletely defined roles in proteostasis, innate immunity, and disease progression; however, investigations into this process have long been hindered by its extremely low abundance and the lack of conformation-specific enrichment tools. Herein, we describe the development of a long-sought conformation-specific antibody, K27-IgG, which can selectively recognize-among all ubiquitin chain types-the unique architecture of K27-linked polyubiquitin (K27-polyUb) characterized by a distinct buried K27-isopeptide bond, with high affinity (KD = 4.66 nM). This antibody was derived from synthetic antibodies initially generated via phage display, using chemically synthesized K27-linked diubiquitin (K27-diUb) as the antigen. High-resolution co-crystal structures uncovered the unique K27-diUb interface targeted by these sAbs. Subsequent reformatting of these sAbs into a full-length human immunoglobulin G (IgG) scaffold yielded K27-IgG, notably exhibiting markedly enhanced affinity without compromising selectivity. Using K27-IgG as a tool, we achieved sensitive detection and immunoprecipitation (IP) of endogenous K27-polyUb in cells, and delineated the intracellular interaction landscape of K27-polyUb through complementary proteomic approaches. Two key findings emerged: 1) The molecular chaperone DNAJB1 is a specific reader of K27-linked ubiquitin chains (but not other linkages) and that K27-polyUb chains themselves exhibit chaperone-like activity, suggesting a novel mechanism by which K27-polyUb regulates chaperone-mediated proteostasis; 2) The E2 enzyme UBE2Q1 assembles K27-diUb, identifying it as a potential writer for this ubiquitin chain topology. Collectively, this study establishes K27-IgG as a robust tool for deciphering the K27-linked ubiquitin code, thereby opening new avenues for investigating the biological functions of K27-linked polyubiquitination.nnHIGHLIGHTS: First K27-linkage conformation-specific antibody with nanomolar affinity overcomes a major barrier in the field.K27-IgG unlocks functional mapping of the K27 ubiquitin landscape under proteotoxic stress.Molecular chaperone DNAJB1 is a selective "reader" of K27-linked ubiquitin chains.K27 chains possess intrinsic chaperone activity, enabling protein refolding and suppressing aggregation.E2 enzyme UBE2Q1 is a "writer" that directly assembles K27-linked ubiquitin chains.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
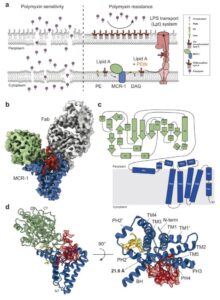
Zinkle A P; Batista M B; Herrera C M; Erramilli S K; Kloss B; Ashraf K U; Nosol K; Zhang G; Cater R J; Marty M T; Kossiakoff A A; Trent M S; Nygaard R; Stansfeld P J; Mancia F
Mechanistic basis of antimicrobial resistance mediated by the phosphoethanolamine transferase MCR-1 Journal Article
In: Nat Commun, vol. 16, no. 1, pp. 10516, 2025, ISSN: 2041-1723.
@article{pmid41298376,
title = {Mechanistic basis of antimicrobial resistance mediated by the phosphoethanolamine transferase MCR-1},
author = {Allen P Zinkle and Mariana Bunoro Batista and Carmen M Herrera and Satchal K Erramilli and Brian Kloss and Khuram U Ashraf and Kamil Nosol and Guozhi Zhang and Rosemary J Cater and Michael T Marty and Anthony A Kossiakoff and M Stephen Trent and Rie Nygaard and Phillip J Stansfeld and Filippo Mancia},
doi = {10.1038/s41467-025-65515-3},
issn = {2041-1723},
year = {2025},
date = {2025-11-01},
urldate = {2025-11-01},
journal = {Nat Commun},
volume = {16},
number = {1},
pages = {10516},
abstract = {Polymyxins are used to treat infections caused by multidrug-resistant Gram-negative bacteria. They are cationic peptides that target the negatively charged lipid A component of lipopolysaccharides, disrupting the outer membrane and lysing the cell. Polymyxin resistance is conferred by inner-membrane enzymes, such as phosphoethanolamine transferases, which add positively charged phosphoethanolamine to lipid A. Here, we present the structure of MCR-1, a plasmid-encoded phosphoethanolamine transferase, in its liganded form. The phosphatidylethanolamine donor substrate is bound near the active site in the periplasmic domain, and lipid A is bound over 20 Å away, within the transmembrane region. Integrating structural, biochemical, and drug-resistance data with computational analyses, we propose a two-state model in which the periplasmic domain rotates to bring the active site to lipid A, near the preferential phosphate modification site for MCR-1. This enzymatic mechanism may be generally applicable to other phosphoform transferases with large, globular soluble domains.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
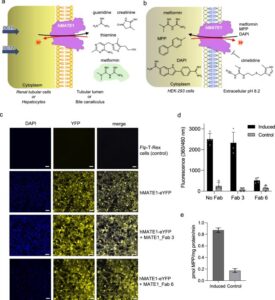
Romane K; Peteani G; Mukherjee S; Kowal J; Rossi L; Hou J; Kossiakoff A A; Lemmin T; Locher K P
Structural basis of drug recognition by human MATE1 transporter Journal Article
In: Nat Commun, vol. 16, no. 1, pp. 9444, 2025, ISSN: 2041-1723.
@article{pmid41145429,
title = {Structural basis of drug recognition by human MATE1 transporter},
author = {Ksenija Romane and Giulia Peteani and Somnath Mukherjee and Julia Kowal and Lorenzo Rossi and Jingkai Hou and Anthony A Kossiakoff and Thomas Lemmin and Kaspar P Locher},
doi = {10.1038/s41467-025-64490-z},
issn = {2041-1723},
year = {2025},
date = {2025-10-28},
urldate = {2025-10-01},
journal = {Nat Commun},
volume = {16},
number = {1},
pages = {9444},
abstract = {Human MATE1 (multidrug and toxin extrusion protein 1) is highly expressed in the kidney and liver, where it mediates the final step in the excretion of a broad range of cationic drugs, including the antidiabetic drug metformin, into the urine and bile. This transport process is essential for drug clearance and also affects therapeutic efficacy. To understand the molecular basis of drug recognition by hMATE1, we determined cryo-electron microscopy structures of the transporter in complex with the substrates 1-methyl-4-phenylpyridinium (MPP) and metformin and with the inhibitor cimetidine. The structures reveal a shared binding site located in a negatively charged pocket in the C-lobe of the protein. We functionally validated key interactions using radioactivity-based cellular uptake assays using hMATE1 mutants. Molecular dynamics simulations provide insights into the different binding modes and dynamic behaviour of the ligands within the pocket. Collectively, these findings define the structural basis of hMATE1 substrate specificity and shed light on its role in drug transport and drug-drug interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}